Single Cell Data Science Core
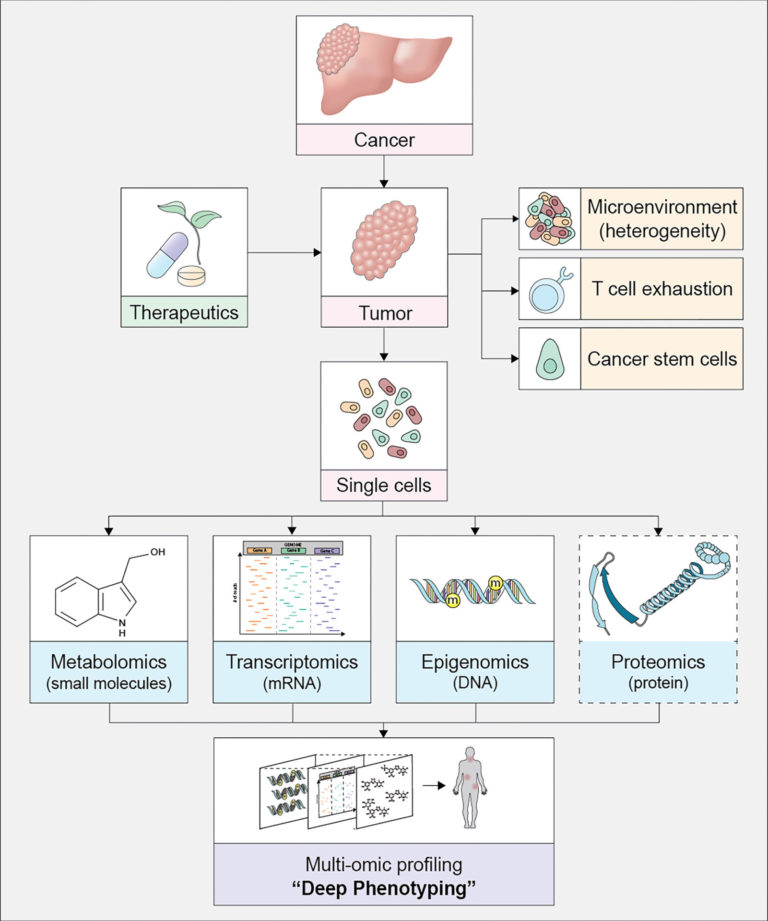
Single cell technologies such as single-cell RNA-seq (scRNA-seq) are rapidly revolutionizing a wide range of biomedical research areas. Unlike traditional bulk RNA-seq technologies that measure global gene expression averaged over a heterogeneous cell population, scRNA-seq examines steady state DNA transcription in individual cells and is thus useful for unveiling transcriptomic heterogeneity at the single-cell level. Life scientists are facing substantial statistical and computational challenges in analyzing single cell data because they are massive, sparse, heterogeneous, and noisy. At the same time, data complexity is growing exponentially. Thus, data must be carefully managed, analyzed, and ultimately made available for discovery, allowing data to be the driving force behind discovery. The complexity of single cell data and its large volume have made it essential to implement novel statistical machine learning methods, as well as quantum computing algorithms for situations when an exhaustive search is required.
To facilitate the analysis of single-cell data, The Texas A&M Regional Center for Excellence in Cancer (TREC) has established a Single-Cell Data Science (SCDS) Core at TAMU. Drs. James Cai, Yang Ni, and Robert Chapkin, along with collaborators, are developing novel statistical methods to address the challenges in:
- Identifying cell types and their subsets,
- Characterizing cellular dynamics,
- Elucidating cell-cell communication and gene regulation at the single-cell level,
- Monitoring structural, functional, or phenotypic changes under different experimental conditions,
- Relating rare transitional cells or cell phenotypes to disease progression,
- Analysis of spatial transcriptomics, and,
- Integration of multi-omics single-cell data. The unique challenge of data integration in single-cell multi-omics is that each observation/cell can be assayed only by one modality. Therefore, neither horizontal nor vertical data integration applies here. This line of research has been supported by CPRIT RP230204 and R01GM148974-01.
Core Director

Professor, Department of Veterinary Integrative Biosciences
Our research focuses on elucidation of genotype-phenotype relationships using computational and evolutionary genomics approaches. We develop computational tools and statistical tests to estimate key parameters of evolutionary processes that shape the sequence and expression variability within and between individuals, populations, and species. We apply theories to genomic diversity and divergence data in order to search for the signatures of selection in the genomes of different organisms.
Core Director

Professor, Department of Veterinary Integrative Biosciences
I am an Associate Professor in the Department of Statistics, Texas A&M University. I am the Co-Director of the Single Cell Data Science Core, a Research Affiliate at the Texas A&M Institute of Data Science, and the Co-Director of the Center for Statistical Bioinformatics. I am interested in developing novel statistics and machine learning methods for Causal Discovery, Machine Learning, Graphical Models, and Bayesian Nonparametrics with applications to cancer multi-omics, HIV electronic health records, and mental health. My research is well-funded by National Science Foundation, National Institutes of Health, National Academies of Sciences, Engineering, and Medicine, and Cancer Prevention and Research Institute of Texas.

The Core
The mission of single-cell data science is to provide bioinformatics and informatics research services that have a measurable impact on the ability of research investigators at Texas A&M University to share their findings and publish their work. Single-cell data science has merged scientific experience with technical know-how to tackle basic research and clinical projects that require skills in single-cell data management, processing, and analytics.
Spatial transcriptomics data analysis
Spatial transcriptomics is a rapidly growing field that promises to characterize tissue organization and architecture at single-cell resolution comprehensively. Single-cell data science provides data analysis service for spatial transcriptomics, which is a powerful approach for TREC cancer research projects, enabling investigators to obtain a holistic understanding of cells in their specific context. For this purpose, we have developed STGEATOOL, Spatial Transcriptomic Gene Expression Analysis Tool, to allow users with limited coding experience to conduct analysis through user interfaces. For example, it enables users to choose any gene of interest and display its spatially resolved expression on the original tissue section. Given that the whole transcriptome is measured, users can choose any number of genes in any combination to view and analyze at the same time. Knowledge of relative locations of cells and gene expression profiles helps to characterize the local cellular environment within the tissue and to better understand the role of different cell types in normal function and tumor pathology.
Multi-omics single-cell data integration and analysis
An integrated analysis of multi-omics single-cell data can generate new knowledge and hypotheses that cannot be otherwise obtained with data collected from any single modality. One of the primary analytic challenges arises when each cell is assayed on a single modality and matching samples across modalities is impeded. To address this challenge, single-cell data science provides cutting-edge single-cell data integration tools that allow joint analyses of multi-omics single-cell data. For example, we have recently developed integrative clustering algorithms that identify cell subtypes of human lymphoblastoid cells using scRNAseq gene expression data and scATAC-seq chromatin accessibility data. The algorithm automatically identifies marker genes of each cell subtype. Our analysis can detect uncharacterized cell subtype structure, leading to the finding of a more heterogeneous nature among lymphoblastoid cells than previously thought. These new approaches can allow TREC investigators to gain a more comprehensive understanding of tumor heterogeneity in a less unbiased, data-driven fashion as compared to traditional bulk multi-omics data and single-modal single-cell data analyses.
Virtual gene knockout analysis
Single-cell data science advanced machine learning tools allow virtual knockout (KO) analysis to be performed with scRNAseq data. Gene KO experiments are a proven, powerful approach to study gene function. However, systematic KO experiments targeting many genes are usually prohibitive due to limited experimental and animal resources. Our method scTenifoldKnk is an efficient virtual KO tool that enables systematic KO investigation of gene function using scRNAseq data. In scTenifoldKnk analysis, a gene regulatory network (GRN) is first constructed with data from wild-type samples, and a target gene is then virtually deleted from the constructed GRN. Manifold alignment is used to align the resulting reduced GRN to the original GRN to identify differentially regulated genes, which are then used to infer target gene functions in analyzed cells. ScTenifoldKnk virtual KO analysis can recapitulate the main findings of real-animal KO experiments and recover gene(s) expected functions in relevant cell types. Systematic virtual KO analysis can be applied to virtually knock out a large number of expressed genes individually to obtain a perturbation profile for each gene. The perturbation profile can be used to infer the function of the KO gene, to identify genes that have similar perturbation profiles, to identify genes that tend to be perturbed together by the same KO gene, and to identify genes involved in specific expression programs characterized in malignant vs. nonmalignant cells.

Impact
Single-cell data science brings together the Data Sciences and Life Sciences communities at TAMU in a novel way. Specifically, the core complements and improves traditional hypothesis-driven research by flipping the model for Cancer Biology and Life Sciences research around so that data drives the experiments leading to novel hypotheses. The core values of single-cell data science include bioinformatics and advanced computation to augment the ability to achieve data science solutions-driven approaches to life science problems and to maintain the strong informatics skill-set necessary for unraveling and integrating data sets originating from various single-cell sources and technologies.
Novelty
Latest publications
Joint Bayesian estimation of cell dependence and gene associations in spatially resolved transcriptomic data | Nature Scientific Reports